INTRODUCTION
Neuromyelitis optica spectrum disorders (NMOSD) is an inflammatory demyelinating central nervous system (CNS) disorder distinct from multiple sclerosis (MS) [1].
The peripheral nervous system (PNS) has not been known to be involved in MS or NMOSD. However, a recent study has reported peripheral neuropathy and chronic inflammatory demyelinating polyneuropathy (CIDP) in patients with MS [2]. In contrast to MS, there have been limited reports on the characteristics of peripheral neuropathy as a complication of NMOSD. Here we report a case of NMOSD that initially presented as left leg weakness and radiculopathy.
CASE REPORT
A 31-year-old woman initially presented with left leg weakness and numbness that had started 2 months ago. Weakness and numbness occurred at the same time. As leg weakness got worse, foot drop occurred one month after developing the numbness. She had visited a local medical center and received lumbosacral spine magnetic resonance imaging (MRI) and electromyography (EMG). However, there was no evidence of peripheral neuropathy on EMG study except reduced recruitment pattern of L5 myotome. Lumbosacral MRI revealed no anatomical abnormalities, suggesting radiculopathy. After that, her left leg weakness was aggravated with gait disturbance. She had no bowel, bladder, or visual symptoms. Past medical and familial history revealed nothing special. Initial neurologic examination revealed normal cranial nerve function. There was weakness of left hip extensor, knee flexor/extensor (extended Medical Research Council grade 4), ankle dorsi-flexor/plantar-flexor, and big toe extensor (grade 3). She had mildly decreased pain, temperature, touch, joint position, and vibration sensations in her left l5 dermatome. All deep tendon reflexes were normal except an absent left ankle jerk. Babinski sign was unclear in her left foot. Testing of the spasticity of her four extremities revealed a Modified Ashworth Scale of zero. She had abnormal gait pattern with genu recurvatum during the standing phase, hip hiking with circumduction, and foot drop during the swing phase.
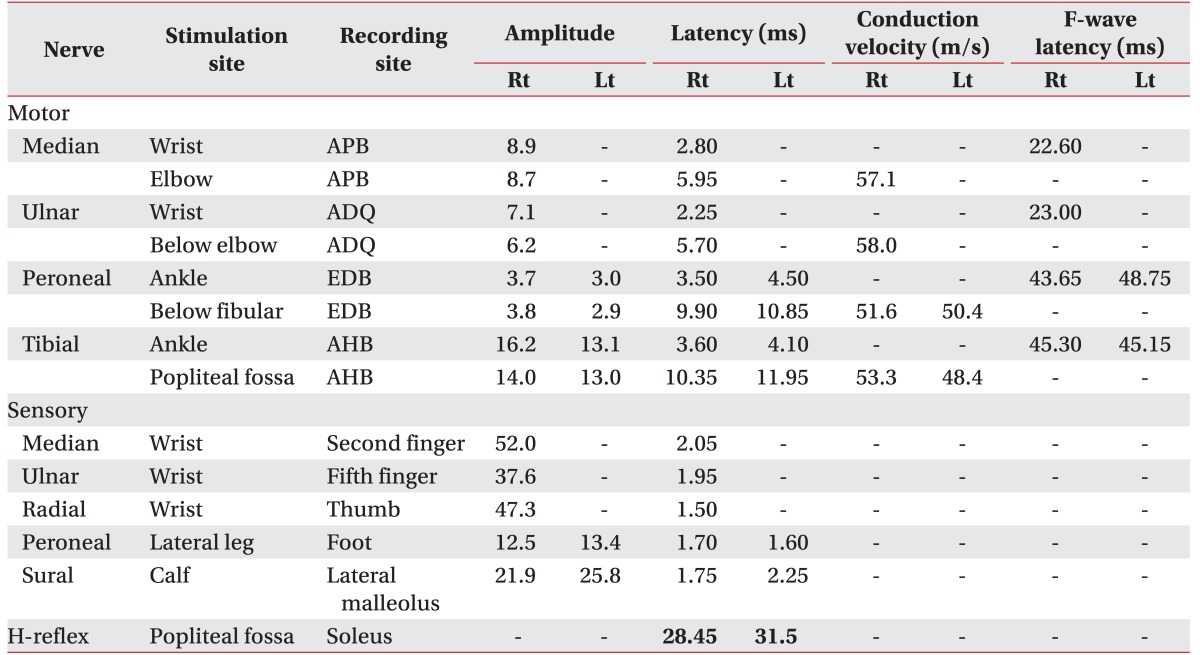
Routine nerve conduction study was normal. F-wave latencies were also normal (Table 1). However, the H-reflex of the left tibial nerve (31.5 ms) was prolonged compared to the right one (28.5 ms). Needle EMG showed active denervation in the left anterior tibialis, tensor fascia lata, and L5 paraspinalis with reduced recruitment in the left tibialis anterior, peroneus longus, extensor halluces longus, gastrocnemius, and tensor fascia lata (Table 2). Because the nerve conduction study with needle EMG findings were not correlated with previous normal lumbosacral MRI finding and there was uncertain Babinski reflex, we measured the sensory evoked potentials (SEPs), motor evoked potentials (MEPs), and visual evoked potentials (VEPs). There were abnormalities in the MEPs (prolonged latencies from left median and tibial nerves). However, SEPs were normal. VEPs showed left P100 latency delay (118.20 ms) compared to the right side (102.40 ms).
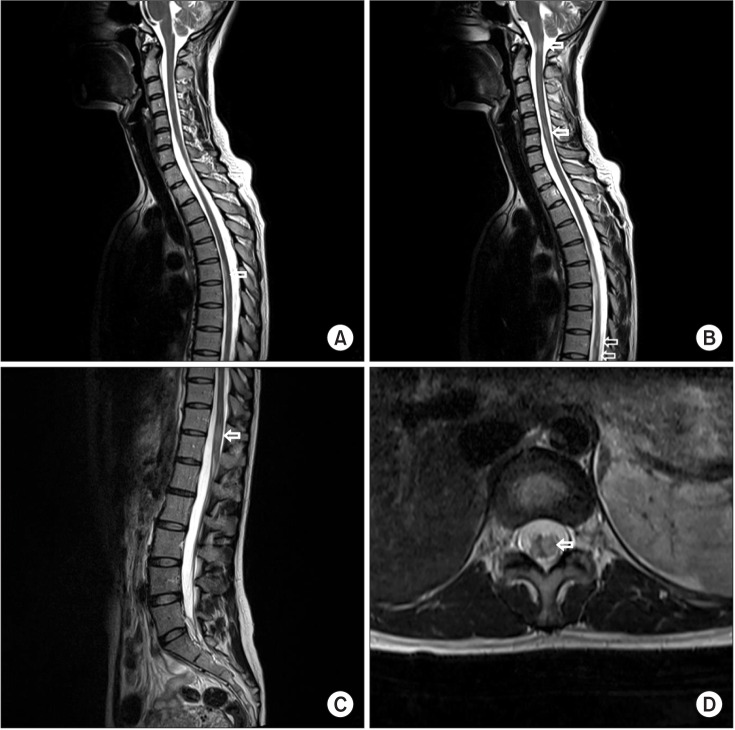
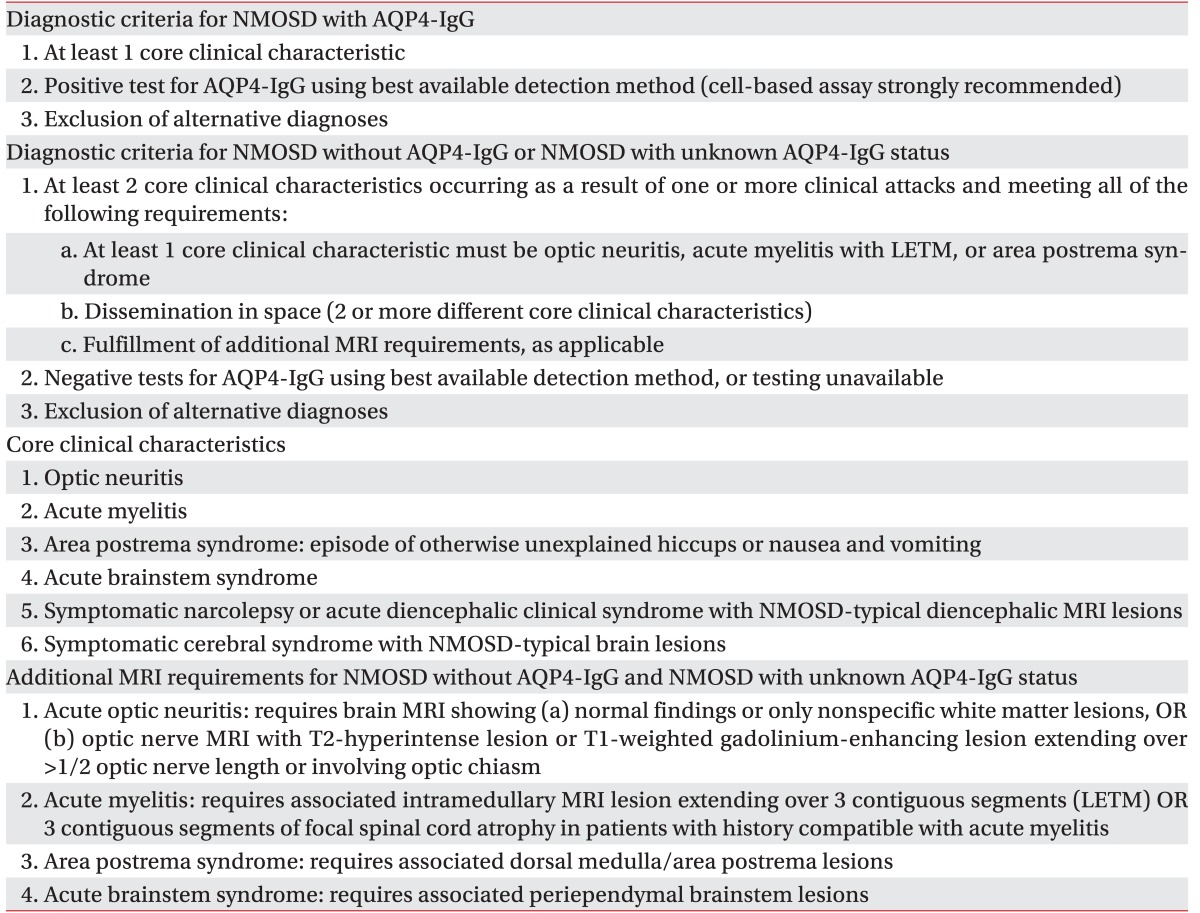
We performed a gadolinium contrast whole spine MRI to evaluate not only other causes of radiculopathy such as ischemia, infection, inflammation but also CNS lesion. This MRI showed multifocal left eccentric T2-weighted high, T1-weighted iso-intensity, and a well-enhanced spinal cord lesions (Fig. 1). Because lesions were limited to spinal cord without spinal root lesion including contrast enhancing imaging, we ruled out radiculitis and conducted gadolinium contrast brain MRI, blood, and cerebrospinal fluid (CSF) examination for CNS disease. Contrast brain MRI revealed multifocal nodular lesions in both periventricular and subcortical white matter, corpus callosum, left dorsal midbrain/pons, right middle cerebellar peduncle, left cervicomedullary junction, and left cerebellar hemisphere (Fig. 2). Routine blood examination had no definite abnormal findings. Viral marker (hepatitis virus B, C, and human immunodeficiency virus), rapid plasma regain titer, rheumatoid factor, lupus anticoagulant, fluorescent antinuclear antibody titer, anti-neutrophil cytoplasmic antibody titer, angiotensin converting enzyme, and anti-glycolipid antibodies (GM1, GM2, GM3, GD1a, GD1b, GD3, GT1b, GQ1b, and Gal-C) were all within normal ranges except GD1b. CSF examination revealed a white blood cell count of 11/µL with 80% lymphocytes. CSF immunoelectrophoresis revealed elevated immunoglobulin G (IgG) levels (5.76 mg/dL; serum IgG, 1,220 mg/dL) and positive oligoclonal bands. Because she is an Asian and contrast whole spine MRI showed spinal cord lesions longer than 3 spinal segments Known as longitudinally extensive transverse myelitis (LETM) lesion, we decided to test aquaporin-4 (AQP4) antibody by indirect immunofluorescence on the recommendation 2010 McDonald criteria for multiple sclerosis [3]. AQP4 antibody test was positive. Finally, we ruled out diseases such as MS, sarcoidosis, and systemic lupus erythematosus and diagnosed her as NMOSD with AQP4-IgG categorized by 2015 NMOSD diagnostic criteria of the International Panel for NMO Diagnosis (IPND) revision [1].
The patient was treated with steroid pulse therapy. Her left leg weakness was improved slightly and treatment was changed to low dose steroid and azathioprine for secondary prevention. Seoul Neuropsychological Screening Battery-2 was performed for evaluating brain lesion effect and newly developing vision problem. Test result revealed decreased visuospatial (6.50%ile) and memory (14.32%ile) function. However, ophthalmic examination was within normal range. Four months later, right lower extremity numbness occurred and new active spinal cord lesion at thoracic level was detected. She was treated with steroid pulse therapy again and her medication was changed to low dose steroid mycophenolate mofetil. Recently, her symptoms wax and wane.
DISCUSSION
As IPND was revised, NMOSD including traditional NMO and modern NMOSD such as opticospinal MS that is more common in Asian countries than in Western countries is classified according to serologic testing (NMO with or without AQP4-IgG). The diagnostic criteria for NMO with or without AQP4-IgG are also different from each other [1] (Table 3). Treatment for NMOSD is divided into treatment of acute attacks and prevention of attacks. Treatment of acute attacks is comprised of intravenous methylprednisolone and plasma exchange. Intravenous methylprednisolone is delivered daily for 3–5 days. If the symptom does not improve, plasma exchange is administered in general. Prophylactic therapy consists of immunosuppressive agents as azathioprine, mycophenolate, and rituximab with or without low dose oral prednisolone [4].
Grey matter and PNS lesions have been reported in CNS demyelination disease such as MS and NMOSD. Compared to MS, there are fewer articles about extra white matter lesions in NMOSD. Kitada et al. [5] have published a case report of NMOSD associated with demyelinating polyradiculoneuropathy. Warabi et al. [2] have reported that one of 28 patients with NMOSD has developed demyelinating polyradiculoneuropathy that does not fulfill the criteria for CIDP without other underlying causes. Feyissa et al. [6] have reported acute axonal polyradiculoneuropathy in seronegative NMOSD. Takai et al. [7] have published two case reports of NMOSD with lumbosacral myeloradiculitis. They not only found demyelination of cauda equina at thoracic 12–lumbar 1 levels in patients, but also expression of AQP4 at the transitional zone of CNS-PNS. These authors argued that transitional zone could be the target of radiculitis in NMOSD. Mingbunjerdsuk and Katirji [8] have reported a case of acute cervical segmental denervation similar to anterior horn cell loss in NMOSD. They suggested that NMOSD proceeds with a similar pathological grey matter and axonal injury to MS due to axoglial antigen as a target of immune attack. Our case is different from other cases except the case reported by Mingbunjerdsuk and Katirji [8] in the aspect of pathophysiology. Our patient's electrodiagnostic finding revealed mainly axonal loss. Her contrast spine MRI showed no definite abnormal finding at the spinal root level. This is different from the report of Takai et al. [7] but with similar pattern to the report of Mingbunjerdsuk and Katirji [8]. Although the accurate pathophysiology of axonal loss in NMOSD is currently unknown, we suggest that axoglial antigen might be a target for immune attack. Our case showed that PNS damage could break out at the early course of NMOSD. Other pathologies besides demyelination might be involved in PNS damage in NMOSD.
In conclusion, NMOSD has various clinical findings. If patients with radiculopathy have no structural abnormality on lumbosacral MRI, demyelinating CNS disease should be taken into consideration. Because PNS could be damaged during early course of NMO, CNS imaging and electrodiagnostic studies as well as CSF examinations should be properly performed if NMOSD is suspected. Early diagnosis and management of NMOSD will have positive effects on patients' long-term prognoses.