Adrenomyeloneuropathy Presenting With Adrenal Insufficiency
Article information
Abstract
Adrenomyeloneuropathy (AMN), one of the variants of X-linked adrenoleukodystrophy (ALD), is inherited peroxisomal disorder associated with the accumulation of very long chain fatty acids (VLCFA). AMN is characterized primarily by involvements of long ascending and descending tracts of the spinal cord and peripheral neuropathy, which leads to spastic paraparesis and urinary and erectile dysfunction. We experienced the AMN case of a 33-year-old man presenting bilateral progressive spastic paraparesis, impotence and urge incontinence with primary adrenal failures, as confirmed by increased serum of VLCFA concentrations. Considering that somatosensory evoked potentials in posterior tibial nerve was the only abnormal finding in electrophysiologic findings when compared with the severe spastic gait pattern shown, it is necessary to follow up with electrophysiologic studies.
INTRODUCTION
X-linked adrenoleukodystrophy (ALD) is an inherited peroxisomal disorder caused by mutation of the ABCD1 gene that results in accumulation of saturated very long chain fatty acids (VLCFA) in plasma, the central and peripheral nervous systems, adrenal glands and testes, which leads to dysfunction of these organs and systems [1]. Adrenomyeloneuropathy (AMN) is a variant of ALD, which affects about 1 in every 20,000 males, it begins in adolescence or early adulthood. The primary manifestation is spinal cord dysfunctions with progressive stiffness and weakness of the legs (spastic paraparesis), abnormal sphincter control, sexual dysfunction and distal polyneuropathy [2]. In Korea, there were several case reports on AMN and some of these were confirmed by serum VLCFA but they all presented normal adrenal functions. This is a case report about AMN as a cause of primary adrenal failures, and confirmed by increased serum VLCFA.
CASE REPORT
In January 2012, a 33-year-old man visited the Department of Rehabilitation Medicine in Seoul Medical Center with complaints of a 13-year history for bilateral, progressive weakness and spasticity in his lower extremities as well as gait disturbance, urge incontinence and impotence. He showed scissoring gait and dragged his feet during walking. He had no family history of neurological or autoimmune disorders and absences of any trauma to the spine. At the age of 15, he was diagnosed with primary adrenal insufficiency so-called Addison disease and has been taking daily Prednisolone of 5 mg.
Physical examinations showed hyperpigmentation, especially in gingiva, tongue, creases of hand, and areolae (Fig. 1). The respiratory, cardiovascular, and abdominal examinations were unremarkable but the muscle bulk in his lower extremities was diminished. On neurologic examination, there was decreased vibratory, pain and temperature sensation below the nipple. In a manual muscle test, flexion and extension of the hip and knee joint each showed 4/4, the ankle dorsiflexion III/III, and the ankle plantarflexion IV/IV under the Medical Research Council classification. He showed spastic gait pattern, with hyperactive deep tendon reflexes and positive Babinski sign. Each degree of spasticity of knees and ankles measured with Modified Ashworth Scale was grade 1. Intellectual and memory functions were within the normal range.

The patient shows hyperpigmentation in gingiva (A), tongue (B), creases of hand (C), and areolae (D).
In laboratory findings, adrenocorticotropic hormone (ACTH) level was above 1,500 pg/mL (normally 10.60 pg/mL) and a short synacthen test showed adrenal failures with a baseline cortisol 0.9 µg/dL (normally 2.9.19.4 µg/dL), 0.7 µg/dL at 30 minutes and 0.6 µg/dL at 60 minutes. The serum testosterone level was 2.40 ng/mL (normally 2.41.8.17 ng/mL). VLCFAs were measured in the plasma. C26:0 levels were 2.969 µmol/L (normally <1.31 µmol/L), the C26:0/C22:0 was 0.083 µmol/L (normally <0.023 µmol/L), and the C24:0/C22:0 was 1.727 µmol/L (normally <1.39 µmol/L) (Table 1).

Very long chain fatty acid (VLCFA) level in patient and reference value
In electophysiologic studies, somatosensory evoked potentials (SEPs) showed no responses in posterior tibial nerve but normal in median nerve. Brainstem auditory evoked potentials (BAEPs) and visual evoked potentials (VEPs) did not demonstrate definite abnormal findings. Nerve conduction studies (NCS) and needle electromyographic examinations were within normal range.
Magnetic resonance imaging showed intramedullary signal changes along entire thoracic cord, which were suggestive of myelopathy (Fig. 2). The diagnosis of AMN was confirmed by raising circulating concentrations of VLCFA.

T-spine magnetic resonance imaging shows a T2-weighted sagittal image of intramedullary signal changes along entire thoracic cord, which are suggestive of myelopathy.
DISCUSSION
The prevalence of adrenal insufficiency is known to every 5 of 10,000 among the general population [3]. Most common cause is the autoimmune disease accounting for 70% to 90%. The remainder consists of other infectious adrenalitis, metastatic cancer, adrenal hemorrhage/infarction, drugs, and inherited disorder such as ALD or AMN (Table 2) [4]. In Korea, tuberculosis is the most common cause of primary adrenal insufficiency [5]. ALD has various phenotypes, including childhood cerebral ALD, adolescent cerebral ALD, AMN, adult cerebral ALD, Addison disease only, and an asymptomatic group. AMN is the most common form of X-ALD and all patients with X-ALD whom reach adulthood develop AMN. ALD begins in the posterior parieto-occipital regions and is often rapidly progressive. The classic form, childhood cerebral ALD, onsets in childhood between the ages of 4 and 6, with rapidly progressive cerebral demyelination leading to spastic tetraparesis, dementia, seizure, visual and auditory dysfunction, although the clinical features may also become manifest in adolescence or adulthood. In contrast, AMN are presented later in life, usually in the third or fourth decade (mean age of onset is 28±9 years) and is primarily characterized by involvements of long ascending and descending tracts of the spinal cord and peripheral neuropathy due to demyelination, which leads to spastic paraparesis and urinary and erectile dysfunction [1]. Approximately two-thirds of male patients have overt or subclinical adrenal insufficiency and a significant proportion have associated gonadal dysfunctions [6]. In our case, adrenal insufficiency was the initial feature and the neurologic symptoms appeared 5 years later. The neurologic abnormalities may appear before adrenal insufficiency, but up to 60% of patients have no or few neurologic symptoms at the time of diagnosis of adrenal insufficiency [7]. Therefore, preclinical AMN should be considered in patients with adrenal insufficiency.
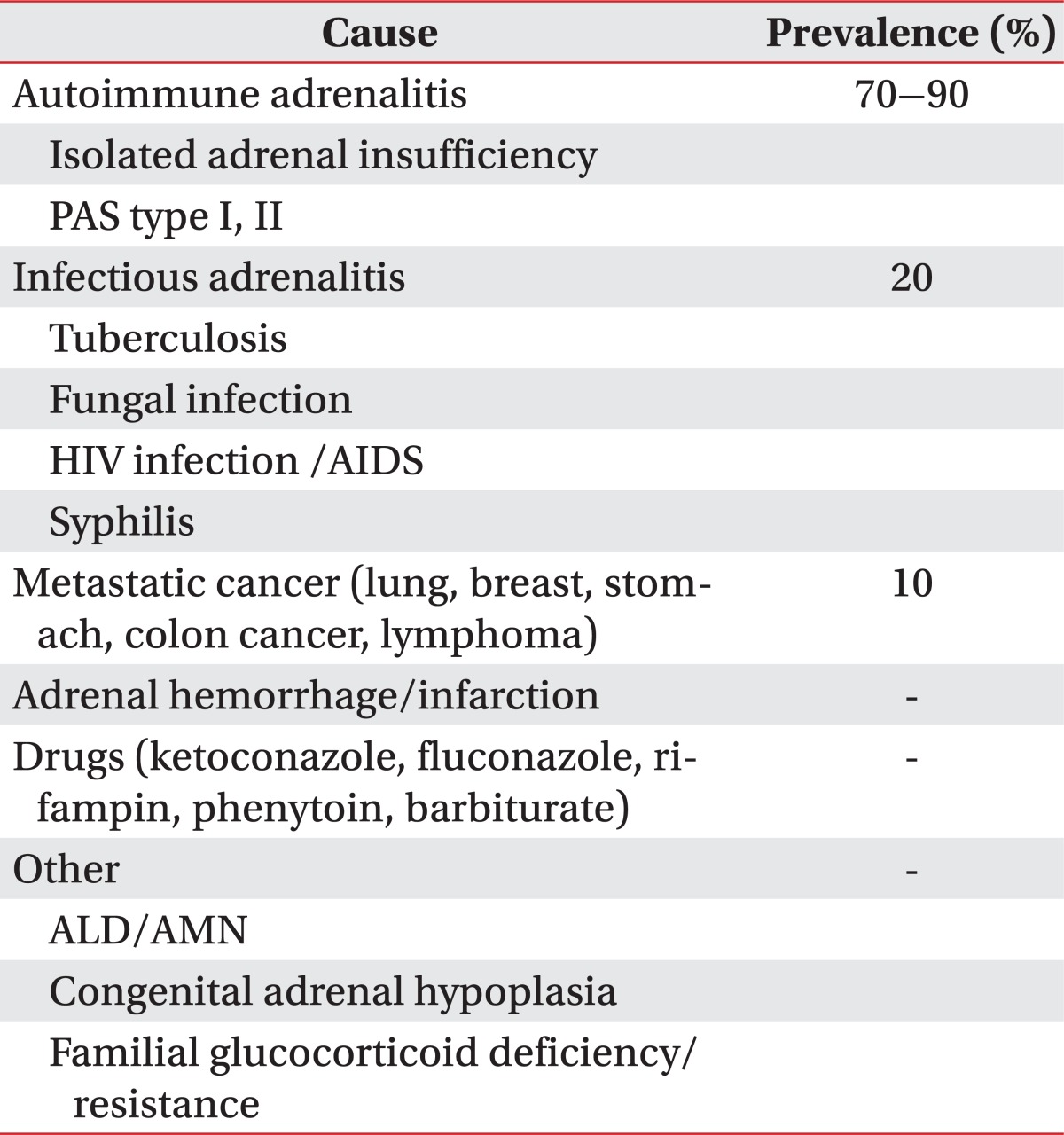
Causes of primary adrenal insufficiency
In laboratory testing, AMN has increased the level of three VLCFA parameters: the level of hexacosanoic acid (C26:0), and the ratio of hexacosanoic acid to tetracosanoic acid (C26:0/C24:0), and to docosanoic acid (C26:0/C22:0). Genetic examination of the ABCD1 gene locus is also important for confirmation of AMN. Adrenal function should be evaluated by plasma ACTH levels and short synacthen tests that identify the rise in plasma cortisol concentration following ACTH stimulation.
In electrophysiologic studies, patients with AMN are associated with a superimposed sensorimotor polyneuropathy. Sensory NCS can be normal but usually reveal mild prolongation in distal latency, reduction in amplitude, and slow conduction velocities [8]. The abnormalities of NCS are more common in males than in females and in legs than in arms, and patients with longstanding diseases usually present more severe abnormalities in nerve conduction parameters than those with recently diagnosed diseases [9]. SEP, particularly of the lower limbs may show subtle abnormalities in the early stage, in contrast, the VEP tend to remain normal. In our case, SEP showed abnormality (no response) in posterior tibial nerve but normal in median SEP, BAEP, and VEP. It may be due to that current findings reflected in the early stage of disease.
Diets low in VLCFAs and supplemented with Lorenzo's oil (erucic and oleic acids) have been suggested to stabilize the disease for mildly affected patients. Lorenzo's oil reduces the synthesis of VLCFA by competitive inhibition of the enzymes responsible for elongation of saturated fatty acids. Lovastatin and sodium phenylacetate normalizes VLCFA level, and may be possible therapeutic agents. Some have advocated bone marrow transplantation and hematopoietic stem cell gene therapy in the treatment of patients with early ALD or AMN [10].
There were some case reports on ALD in Korea but mostly childhood cerebral form and previous case reports about AMN dealt with patients presenting normal adrenal functions. No studies on AMN presenting adrenal insufficiency confirmed by increasing VLCFA were reported in Korea. So, we assumed that it might be the first report on AMN as a cause of primary adrenal insufficiency. Although SEP in posterior tibial nerve was only in abnormal findings of electrophysiologic studies, it is suggested that abnormal SEP in median nerve and NCS findings would be evoked at follow-up studies.
Notes
No potential conflict of interest relevant to this article was reported.