A Nine-Month-Old Boy With Isodicentric Chromosome 15: A Case Report
Article information
Abstract
Isodicentric chromosome 15 [idic(15)] is a rare chromosomal abnormality that occurs due to inverted duplication of chromosome 15q. It is associated with many clinical findings such as early central hypotonia, developmental delay, cognitive dysfunction, autism spectrum disorders, and seizure. Delayed development is a common problem referred to pediatric rehabilitation clinics. A 9-month-old boy with delayed development was referred to our clinic for assessment and treatment. On chromosomal analysis, he was diagnosed as idic(15) syndrome with 47,XY,+idic(15)(q12) on karyotyping. Herein we describe his clinical manifestations and provide a brief review of the related literature.
INTRODUCTION
The chromosome region 15q11-q13 is known for its instability and is highly susceptible to genomic rearrangements [1]. Many rearrangements may occur in this imprinted segment such as deletions associated either with Angelman syndrome (AS) or Prader-Willi syndrome (PWS) according to parental origin; these involve translocations, inversions, and supernumerary marker chromosomes formed by the inverted duplication of chromosome 15 [Inv dup(15)] [2]. Inv dup(15) syndrome is the most common of the heterogeneous group of extra-structurally abnormal chromosomes [3].
Inv dup(15) or isodicentric chromosome 15 [idic(15)] displays distinctive clinical findings represented by hypotonia, developmental delay, mental retardations, difficulty with controlling epilepsy, autism, and autistic like behavior. It can thus be clinically suspected prior to cytogenetic confirmation [4].
We present a case of idic(15) syndrome in a 9-month-old boy who was referred to our clinic for delayed development. The clinical features of this syndrome in this early period have been rarely reported. In addition, the youngest reported case of idic(15) syndrome was in an 11 months old [5] and no cases of this syndrome have been reported in Korea, to the best of our knowledge. We therefore report a case of idic(15) syndrome and provide a brief literature review.
CASE REPORT
A 9-month-old boy visited our clinic with development. He was born at term by normal delivery with birth weight of 3.7 kg after an uneventful pregnancy. The family history was non-contributory. There were no notable prenatal and immediately postnatal problems. He had been taken to another clinic at four months because he could not hold his head. Brain magnetic resonance image scan, thyroid function test, and metabolic work ups were done at that time with normal results. At nine months old, he visited our clinic due to his persistent developmental problems. Minor craniofacial dysmorphism involving mildly deep-set eyes, downslanting palpebrae, microcephaly, and hypotonic face were observed (Fig. 1). On physical examination, there were no abnormal neurological signs such as persistent primitive reflex, ankle clonus, Babinski's sign or hyperactive deep tendon reflexes. Diffuse hypotonia was noted. He could hold his head at six months and sat with support at nine months, but he could not sit without support nor crawl reciprocally at that time. He could reach with his arm to get a toy with palmar grasp. He was babbling and attentive during play, also was able to share enjoyment, to pleasure with other people, and to make eye to eye contact. Stereotyped movements such as repetitive hand twisting or head turning and hand wringing were not observed. He had a Mental Developmental Index (MDI) score of 74 and a Psychomotor Developmental Index (PDI) score of 51 on Bayley Scales of Infant Development II tests and a social intelligence quotient (SQ) of 79.3 on the social maturity scale. His subcategorical development was estimated at four to five months in the cognitive area, four months in the language and motor areas, and six months in the social areas. On chromosomal study, chromosomal abnormality of 47,XY,+idic(15)(q12) was identified (Fig. 2). Chromosomal analysis for his parents was strongly recommended but this was refused. He had physical and occupational therapy in order to reduce his developmental delay. He sat without support at 11 months, could crawl at 13 months, came to walk along the furniture at 17 months, and stood alone for a few seconds at 19 months. Psychological evaluation at this point showed scores below 50 on both the MDI and PDI of the Bayley scale and the SQ of 68.3 for social maturity. His overall developmental lags were seven months in the social and language areas, and eight to nine months in other areas. On the Childhood Autism Rating Scale (CARS), his score was 15 and he did not show any distinctive autism or autistic like behavior. At 19 months he had no history of clinically notable seizure and his electroencephalography (EEG) findings were normal.
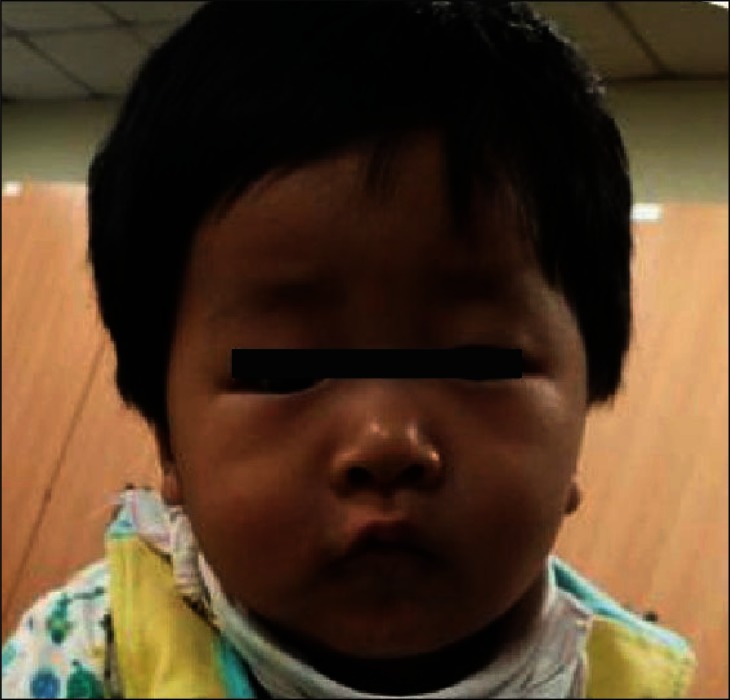
The boy shows some facial dysmorphism including downslanting palpebral fissures, deep-set eyes and microcephaly.
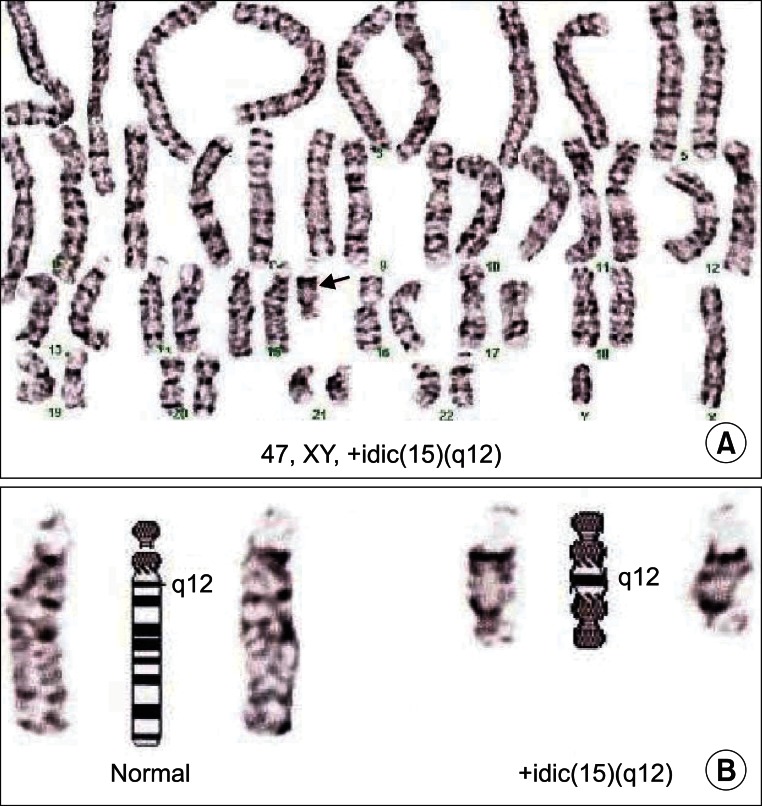
Karyotyping of subject shows isodicentric chromosome 15 [idic(15)]. (A) Idic(15) is indicated by an arrow. (B) Comparison of iIdic(15) with normal chromosome 15. Two centromeres and q12 bands can be seen in Idic(15).
DISCUSSION
Isodicentric 15 syndrome, also called idic(15) or inv dup(15), is a chromosome abnormality that results from duplications of chromosome 15q11.q13. Individuals with idic(15) are typically born with 47 chromosomes, with the extra chromosome being made up of a piece of chromosome number 15 that has been duplicated end to end like a mirror image [3]. The incidence of this syndrome is not fully known but is estimated to be 1 in 30,000 with a sex ratio of almost 1 [4]. Generally this syndrome is not inherited and occurs de novo [4,6].
There is wide range of severity in the developmental problems experienced by individuals with idic(15) syndrome. Various genetic mechanisms have been hypothesized to explain this clinical heterogeneity, including the size of chromosomal duplication, dosage effect of genes in this region and the imprinting mechanism. In most cases, the maternally inherited aberration of chromosome 15q11-q13 seems to be pathogenic [4].
The clinical features of idic(15) syndrome consist of severe developmental delay and intellectual disorder (ID), speech impairment, epilepsy, altered behavior, diffuse hypotonia, and minor craniofacial dysmorphism. Diffuse hypotonia, developmental delay, and intellectual disorder affect all individuals with idic(15). ID has been found to be severe to profound in 85% and mild to moderate in 15% of cases [1,4]. The ages of acquisition of motor milestones are rarely reported in the literature. However it seems that sitting is generally achieved between 10 and 20 months of age and walking between two and three years [4,7]. Many children with idic(15) share similar facial characteristics including a flat nasal bridge, inner epicanthal fold, deep set eyes, low set ears and downslanting palpebrae. The clinical findings of our case seem to be compatible with these findings [1].
Autism or autistic-like behavior has been commonly described in individuals with idic(15) and thus an association between the syndrome and autism is suggested [4]. However, standardized assessment for autism has not been used in most studies of the syndrome. One study attempted to elucidate the true relationship between idic(15) and autism using a standardized assessment tool in 29 individuals with the syndrome. Of these individuals, 20 had a high probability of being autistic and eight of the nine remaining children were under five years of age. The authors suggest that the onset of some autistic features may, as such, be delayed [8]. Our subject did not show any autism or autistic-like behavior up to 19 months, but these findings indicate there is a possibility he may develop autism and autistic behavior at a later point. The natural history of this syndrome in relation to the development of autism is a matter for future longitudinal studies to investigate.
Seizures represent an important medical feature of this syndrome. Over 75% of sufferers have at least one seizure and a wide variety of seizures may occur [4]. In our case, a history of seizures had not occurred at 19 months, but careful monitoring of future developments in this area is warranted.
At the present time, there is no specific treatment that can directly address the genetic changes in these individuals. Symptomatic management associated with idic(15) may be helpful to minimize the consequences of this genetic syndrome.
In conclusion, idic(15) is considered as a clinically recognizable disorder. Clinicians should suspect this syndrome in any infant or child with early central hypotonia, minor dysmorphic features, developmental delay/ID and autism-like behaviors and who subsequently develops hard to control seizures/epilepsy [4]. However, there is a large phenotypic variability and thus it is not always possible to recognize this syndrome through only its clinical features. Chromosomal analysis will be helpful in some cases. Early identification of this syndrome may be useful for providing appropriate and timely intervention through careful attention to the development of seizure and autism or autistic-like behavior. Our study provides information as to the early clinical features of idic(15) seen in our case and provides a literature review.
Notes
No potential conflict of interest relevant to this article was reported.